Genetiske hjertesykdommer
Genetiske hjertesykdommer er en samlebetegnelse for en rekke ulike arvelige hjertesykdommer som kardiomyopatier, inkludert arytmogen høyre ventrikkel-kardiomyopati, hypertrofisk kardiomyopati og dilatert kardiomyopati, og ionekanalsykdommer som lang QT-syndrom, katekolaminerg polymorf ventrikkeltakykardi og Brugadas sykdom.
Graden av symptomer kan variere betydelig hos pasienter med samme diagnose. Felles for sykdommene er at de kan gi økt risiko for hjerterytmefortyrrelser og at de kan debutere med akutte livstruende hendelser som alvorlig hjerterytmeforstyrrelse og hjertestans.
Hos cirka halvparten av alle pasienter med genetiske hjertesykdommer påvises det en sykdomsfremkallende mutasjon, med noe variasjon i andelen mutasjonspositive mellom de ulike diagnosene.
Arvegangen er oftest autosomal dominant. Det betyr at risikoen for at man skal arve mutasjonen fra én av sine foreldre er femti prosent og at den er like stor for gutter og jenter.
Alle de nevnte sykdommene har såkalt variabel penetrans, hvilket betyr at man ikke trenger å være syk selv om man har mutasjonen. For eksempel kan to søsken som begge har arvet mutasjonen ha stor variasjon i symptomer, én kan være alvorlig syk og én kan være helt uten symptomer på hjertesykdom.
I det samme så de trollene komme settende, og de var så store og digre at hodene på dem var jevnhøye med furutoppene. Men de hadde bare ett øye sammen alle tre, og det skiftedes de til å bruke; de hadde et hull i pannen, som de la det i, og styrte det med hånden; den som gikk foran, han måtte ha det, og de andre gikk etter og holdt seg i den første.
Kardiomyopatier
Kardiomyopatier er primære hjertemuskelsykdommer. Diagnosen stilles etter at man har utelukket andre bakenforliggende årsaker som høyt blodtrykk, klaffefeil eller medfødte hjertemisdannelser.
Kardiomyopatier hos barn har svært varierende alvorlighetsgrad. En del barn utvikler alvorlig sykdom med tidlig debut, rask sykdomsprogresjon og dårlig prognose, men andre kun har lette hjertemuskelforandringer, ofte identifisert i forbindelse med familieutredning.
Hypertrofisk kardiomyopati
Ved hypertrofisk kardiomyopati (HCM) ses økt veggtykkelse av hjertemuskelen. Årsaken til HCM hos barn er mer variert enn det man ser hos voksne og inkluderer genetiske syndromer, metabolske sykdommer og nevromuskulære lidelser.
De fleste tilfellene med HCM skyldes imidlertid en sykdomsgivende genfeil (mutasjon) som fører til forandringer i hjertemuskelcellens strukturer som har med hjertemuskelcellens sammentreknings-evne å gjøre (sarkomermutasjoner).
Dersom man tar en vevsprøve fra hjertemuskelen, ser man at hver enkelt muskelcelle er forstørret, og i enkelte områder er hjertemuskelcellene organisert i et uregelmessig mønster. Det er også økt mengde arrvev i hjertemuskelen, noe som sannsynligvis skyldes både redusert oksygentilførsel til hjertemuskelen og økt produksjon av arrvev.
Historisk sett ble sarkomerisk HCM ansett som uvanlig hos barn, men nyere europeiske og nord-amerikanske studier har vist at sarkomerisk sykdom også kan debutere i tidlige barneår.
Oftest er det hjerteskilleveggen mellom venstre og høyre hovedkammer som fortykkes mest. Dersom fortykkelsen sitter i venstre hjertekammers utløp, kan dette føre til at blodstrømmen hindres når blodet pumpes ut i kroppen.
Dette kalles obstruktiv HCM. Hos pasienter med obstruktiv HCM kan man ofte høre en bilyd når man lytter til hjertet med stetoskop.
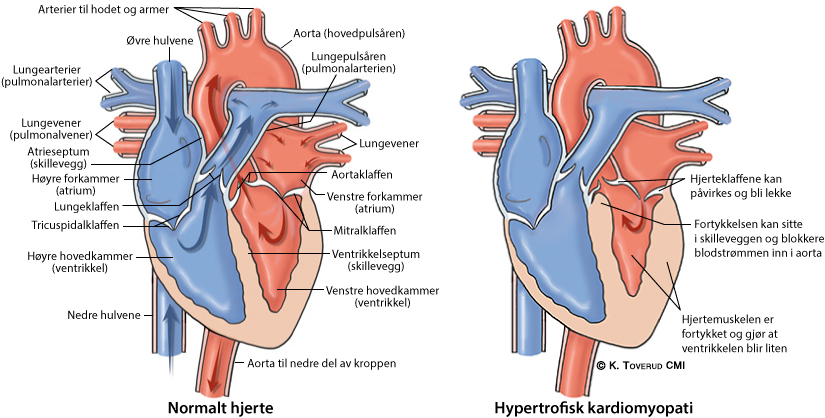
Dersom man kun er bærer av en mutasjon som kan gi HCM, uten å ha utviklet forstørret hjerte, har man som regel ingen symptomer.
Pasienter med moderat eller betydelig grad av fortykket hjertemuskel, og da særlig obstruktiv HCM, blir i varierende grad slitne i forbindelse med anstrengelse, og kan også være plaget med brystsmerter. Pasienter med HCM har økt risiko for ulike rytmeforstyrrelser.
Det kan være aktuelt å starte behandling med betablokker for å redusere hjertefrekvensen og stabilisere hjerterytmen. Hos pasienter med obstruktiv HCM kan det i noen tilfeller også være nødvendig med kirurgisk septal myektomi, et hjertekirurgisk inngrep hvor man opererer bort en liten bit av det mest fortykkede området i hjerteskilleveggen.
Alle pasienter med HCM vurderes med tanke på hvor stor risiko det er for alvorlige hjerterytmeforstyrrelser og om det er behov for implanterbar hjertestarter (ICD).
Vi oppfatter genbærere uten symptomer eller økt tykkelse av hjertemuskelen og uten arytmi-episoder som friske og disse får ingen restriksjoner når det gjelder fysisk trening. Tidligere retningslinjer har anbefalt at pasienter med HCM skal avstå fra konkurranseidrett.
Det foreligger imidlertid nyere forskning som skaper tvil om høyintensitetsaktivitet virkelig er uheldig hos HCM-pasienter. Idrettsutøvere med mild hypertrofi, normal hjertefunksjon og ingen obstruksjon av blodstrømmen ut fra hjertet er ofte i stand til å trene intensivt.
Arytmogen høyre ventrikkel-kardiomyopati
Arytmogen høyre ventrikkel-kardiomyopati (ARVC) kjennetegnes av økt risiko for hjerterytmeforstyrrelser og svekkelse av hjertemuskulaturen, oftest i høyre hjertehalvdel.
ARVC skyldes vanligvis en genfeil som gir endringer i desmosomene i hjertet. Desmosomer er proteiner som binder sammen hjertemuskelcellene med hverandre. Man kan forestille seg desmosomene som limet mellom cellene. Når desmosomene er svekket, vil mekanisk stress, som ved fysisk aktivitet, kunne føre til at limet mellom cellene ryker og hjertemuskelceller går til grunne.
Disse hjertemuskelcellene blir erstattet av fett og bindevevsceller. Når deler av hjertet består av bindevev, er man mer utsatt for hjerterytmeforstyrrelser.
Det mest typiske er at sykdommen manifesterer seg i tidlig voksen alder, men nyere studier, blant annet fra Oslo Universitetsykehus, har vist man kan utvikle alvorlig ARVC også i barnealder. Dessverre kan alvorlig hjertearytmi og hjertestans være det første symptomet på ARVC, og sykdommen er den vanligste årsaken til plutselig død blant unge i Skandinavia.
Svimmelhet og synkope, særlig i forbindelse med fysisk aktivitet, er symptomer man skal være oppmerksom på, da de kan være uttrykk for alvorlig hjerterytmeforstyrrelse. Sykdommen kan også føre til hjertesvikt, og det finnes eksempler på svært unge ARVC-pasienter som har hatt behov for hjertetransplantasjon.
Medikamentell behandling, i form av betablokker, vurderes hos alle pasienter med ARVC for å minske risiko for arytmi. Det kan også bli aktuelt å operere inn en implanterbar hjertestarter. Denne registrerer hjerterytmen og kan avlevere livreddende sjokk ved en eventuell hjertestans.
Vi har lite kunnskap om hvilken betydning fysisk aktivitet har for sykdomsutviklingen hos barn med ARVC, og det gis i utgangspunktet ingen restriksjoner på fysisk aktivitet så lenge det ikke er tegn til hjertesykdom. Studier på voksne ARVC-pasienter har imidlertid vist at høyintensitetsidrett er forbundet med tidligere debut av hjerterytmeforstyrrelser og et mer alvorlig sykdomsforløp.
Høyintensitetsidretter og en idrettskarriere anbefales derfor ikke hos barn med genvarianter forbundet med ARVC.
Dilatert kardiomyopati
Dilatert kardiomyopati er en hjertemuskelsykdom som fører til utvidelse av hjertets kamre og redusert hjertefunksjon.
DCM er den vanligste årsaken til kardiomyopati i barndommen og kan blant annet skyldes infeksjoner, hjerterytmeforstyrrelser som gir vedvarende rask hjerterytme, toksiner (inkludert kjemoterapi), familiær DCM, medfødte metabolske sykdommer og nevromuskulære sykdommer.
Antallet kjente sykdomsfremkallende genvarianter øker. Noen eksempler er lamin A/C (LMNA), titin (TTN), fosfolamban (PLN), RNA-binding motif-protein (RBM20), Bcl- 2-assoisiert athanogen (BAG) og filamin C (FLNC).

Barn med DCM kan debutere med hjertesvikt, med symptomer som varierer fra spisevansker til kardiovaskulær kollaps. Etter spedbarnsalder er DCM den vanligste årsaken til hjertetransplantasjon hos barn. Sykdommen kan også debutere med hjerterytmeforstyrrelse.
Dette gjelder særlig for pasienter med LMNA-mutasjoner hvor det er økt risiko for atrieflimmer, AV-blokk og alvorlig hjertearytmi med hjertestans. Noen av pasientene med LMNA-mutasjon har i tillegg skjelettmuskelsykdom.
Aktuell behandling er medikamenter som reduserer risiko for arytmi, samt hjertesviktmedisiner. Det kan også bli aktuelt med implanterbar hjertestarter (ICD).
Studier på voksne pasienter med DCM viser at moderat trening forbedrer fysisk kapasitet og hjertefunksjon. Det gis i utgangspunktet ingen restriksjoner på fysisk aktivitet hos barn med DCM-relaterte genvarianter så lenge det ikke er tegn til hjertesykdom.
Imidlertid bør man være obs på besvimelser relatert til fysisk aktivitet, da dette kan være uttrykk for en alvorlig hjerterytmeforstyrrelse. Satsing på høyintensitetsidretter og en idrettskarriere anbefales i utgangspunktet ikke.
Å avstå fra konkurranseidrett er særlig viktig for pasienter med sykdomsfremkallende varianter i LMNA, da det er data som tyder på at høyintensitetstrening kan ha en negativ effekt på hjertefunksjonen hos disse.
Ionekanalsykdommer
Hvert hjerteslag er styrt av elektriske impulser i hjertemuskelcellene. Disse elektriske impulsene oppstår ved at ladete partikler (ioner) transporteres gjennom ionekanaler i cellemembranen inn og ut av hjertemuskelcellene.
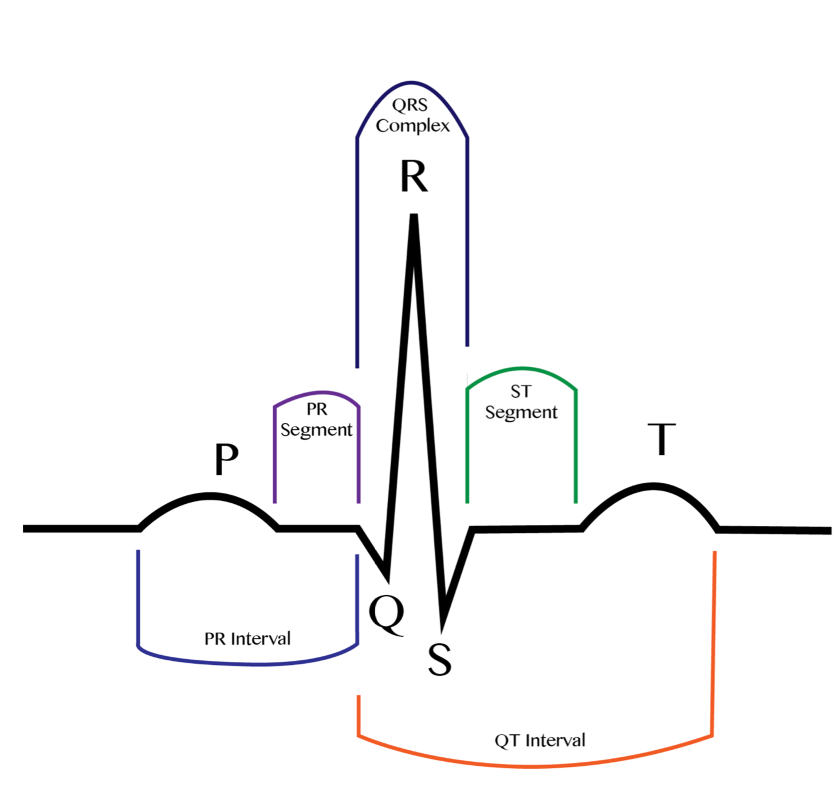
Ved å ta elektrokardiogram (EKG) kan disse elektriske impulsene registreres, og man ser da et bestemt mønster som er angitt med bokstavene P, Q, R, S og T. Hvert QRS-kompleks representerer et hjerteslag.
Ionekanalsykdommer skyldes mutasjoner i gener som koder for ionekanaler. Siden ionekanaler er sentrale elementer i overledningen av elektriske impulser i hjertemuskelcellene, kjennetegnes disse sykdommene av økt arytmirisiko, med plutselig død som en fryktet presentasjonsform.
Lang QT-tid-syndrom (LQTS)
Ved Lang QT-tid-syndrom (LQTS) er avstanden mellom Q og T i EKG lenger enn normalt. Hjerterytmeforstyrrelser preger symptombildet, i form av hjertebank, svimmelhet, besvimelser eller hjertestans.
Mutasjoner i 17 ulike gener er vist å være assosiert med LQTS, men det klart vanligste er mutasjoner i gener som koder for kaliumkanaler. Det er ulike triggere for hjerterytmeforstyrrelser ved de forskjellige gentypene.
Ved LQT1 er det fysisk aktivitet og økt hjertefrekvens som typisk utløser rytmeforstyrrelser. Ved LQT2 er det at man skvetter (f.eks. av vekkerklokke) og emosjonelt stress vanlige triggere. Ved LQT3 er hjerterytmeforstyrrelsene ofte assosiert til lav puls, og de oppstår gjerne ved hvile eller søvn. Hjerterytmeforstyrrelser i forbindelse med svømming forekommer relativt ofte og særlig ved LQT1.
Tiltak for å endre livsstilen er viktig hos pasienter med lang QT. Det viktigste er å unngå QT-forlengende medikamenter (en oppdatert liste finnes på www.crediblemeds.org) og å være nøye med tilstrekkelig væskeinntak og salttilskudd ved mye svetting og ved oppkast/diaré.
Pasienter med lang QT skal aldri bade alene. Barn kan delta i fysisk aktivitet inntil hva de selv orker, men skal ikke presses utover det. Det anbefales å avstå fra konkurranseidrett. Disse rådene gjelder også for familiemedlemmer som har fått påvist LQTS ved gentesting, men som ikke har forlenget QT-tid i EKG.
Betablokker anbefales til alle pasienter med forlenget QT-tid, men skal også vurderes hos de med LQTS uten symptomer på sykdommen. For de fleste er dette tilstrekkelig beskyttelse. I alvorlige tilfeller må det vurderes om man skal operere inn en implanterbar hjertestarter.
Brugada syndrom
Ved Brugada syndrom ser man et bestemt mønster på EKG. Hos noen er dette alltid til stede, hos andre er det kun synlig under bestemte betingelser. Brugada syndrom skyldes mutasjoner i gener som koder for natriumkanaler i hjertet.
Det er lav forekomst av Brugada syndrom i Skandinavia, men tilstanden er hyppigere i deler av Asia.
Pasienter med Brugada syndrom har økt risiko for å utvikle potensielt livstruende hjerterytmeforstyrrelser. Rytmeforstyrrelsene oppstår oftest i forbindelse med søvn og hvile. Overoppheting, store matinntak og bruk av visse medisiner kan også utløse hjerterytmeforstyrrelser.
Det er kun implanterbar hjertestarter som er effektiv behandling ved Brugada syndrom. Viktige livsstilstiltak for å forebygge hjerterytmeforstyrrelser er å unngå overoppheting, gi febernedsettende medisiner ved feber og å være nøye med rehydrering og salttilskudd ved oppkast og diaré. Det er ikke påvist sammenheng mellom fysisk aktivitet og økt risiko, men man bør som nevnt unngå overoppheting,
En rekke medikamenter kan gi økt risiko for hjerterytmeforstyrrelser hos pasienter med Brugada syndrom og disse bør man holde seg unna. Det foreligger en liste på www.brugadadrugs.org som oppdateres jevnlig.
Katekolaminerg polymorf ventrikkeltakykardi (CPVT)
CPVT er en arvelig hjertesykdom som disponerer for hjerterytmeforstyrrelser under fysisk eller psykisk stress. Navnet kommer av at det er stresshormoner (katekolaminer) som utløser hjerterytmeforstyrrelsene (takykardi). Denne startet i hjertets hovedkammer (ventrikkel) og ved rytmeregistrering ser alle hjerteslagene litt forskjellige ut (polymorf).
Årsaken til CPVT er genfeil som påvirker evnen til å styre nivået av kalsium i hjertemuskelcellene. Kalsium er viktig for normal sammentrekning av cellene.
Hjerterytmeforstyrrelsene kan føre til svimmelhet, hjertebank, besvimelser eller hjertestans. Det typiske er at disse symptomene er relatert til fysisk eller psykisk stress. Pasienter med CPVT bør unngå situasjoner med mye psykisk stress. De frarådes å drive med konkurranseidrett og bør ikke bade alene. Betablokker er standard medikamentell behandling. For noen vil det være aktuelt med en implanterbar hjertestarter.
Generelle aspekter
Genetisk kardiologi er et fagområde som har gjennomgått en stor utvikling de siste årene. Antall kjente sykdomsfremkallende mutasjoner og forståelsen av disse har økt raskt, men det er fremdeles mange ubesvarte spørsmål.
Når en pasient får diagnostisert en arvelig hjertesykdom, og man finner en spesifikk mutasjon, vil dette utløse en genetisk kaskadetesting av slektninger. Man får da ofte identifisert flere nye pasienter, og mange av disse har ingen symptomer på diagnosetidspunktet.
Mutasjonspositive familiemedlemmer skal følges av helsevesenet. Noen pasienter med påvist mutasjon kan gå gjennom hele livet uten å utvikle sykdom, mens andre med samme mutasjon utvikler sykdommen i ung alder. Vurdering av hver enkelte pasients risiko for å utvikle alvorlig hjerterytmeforstyrrelse, er særlig viktig. Derfor foregår det mye forskning på dette området.
Familieutredning kan ha store konsekvenser: både psykisk, sosialt, arbeidsmessig, for idrett, forsikringer osv. Det kreves derfor utfyllende informasjon og samtykke før man eventuelt starter en genetisk utredning av asymptomatiske familiemedlemmer.
Hva som er den optimale alder for å starte genetisk screening og klinisk oppfølging av unge familiemedlemmer er ikke tilstrekkelig definert. Det foreligger få studier på barn med genetisk hjertesykdom.
Når det gjelder ionekanalsykdommer, bør testing gjøres allerede ved fødsel. Ved genetiske kardiomyopatier anbefales det at barn gentestes på lik linje med andre familiemedlemmer, uavhengig av alder, og at oppfølgingen individualiseres i forhold til eventuell forekomst av tidlig sykdomsdebut i familien og forventet utvikling av sykdommen.
Barn med genetisk hjertesykdom kan ha økt risiko for hjerterytmeforstyrrrelser. Det er imidlertid viktig at barna opplever minst mulig begrensninger. De bør få være med på aktiviteter på lik linje med de andre og delta i idrett inntil hva de selv orker, men skal ikke presses utover det.
Denne artikkelen er revidert høsten 2024 og skrevet av barnekardiolog Marit Kristine Smedsrud.
Kvalitetssikret og utfyllende informasjon fra eksterne kilder.